 |
1) Load the protein structure:
The first step consists in loading the three-dimensional (3D) structure (experimental or modelled) of the target protein. There are two options : one can either provide the 4-letter code of a structure from the Protein Data Bank (PDB) which is then automatically retrieved, or upload a personal structure file in PDB format. After this step, SCooP provides a brief summary indicating the name of the protein, the different chains, and the number of residues per chain. In case the structure file contains multiple NMR models, only the first model is considered. If the PDB code introduced by the user is not present in the PDB or if the uploaded structure file is not in PDB format, an error message is displayed.
|
2) Choose the correct chain and the host organism:
SCooP is designed to be applied to monomeric chains that undergo a two-state (un)folding transition. Therefore, once the PDB is uploaded and the chains are displayed, the user must select one of them. Moreover, when the structure file has been manually uploaded, the user is asked to select the name of the host organism from a list. After this step, the computation process starts.
|  |
Upon submission of the query, SCooP provides a "Job ID" and a link that must be followed/bookmarked to check the status of the job. The results are available on this page as soon as the computations are over (you may need to refresh the page). The results remain on the webserver for two weeks.
To predict the stability curve of the target protein, SCooP uses temperature-dependent statistical potentials, derived from sets of mesostable, thermostable and reference proteins. The potentials are of two types : backbone torsion angle potentials that describe local interactions along the polypeptdide chain and distance potentials that describe tertiary interactions. SCooP also utilizes the total number of residues of the chain, the temperature of the host organism (that are stored in a manually curated dataset) and the total accessible surface area of the structure.
The results are reported in the Curve pages. They consist in the plot of the full Gibbs-Helmholtz curve, the equation of the curve, and the predicted values of all the thermodynamic quantities that characterize the folding transition. A pdf file containing these results is also generated and can easily be downloaded by the user.
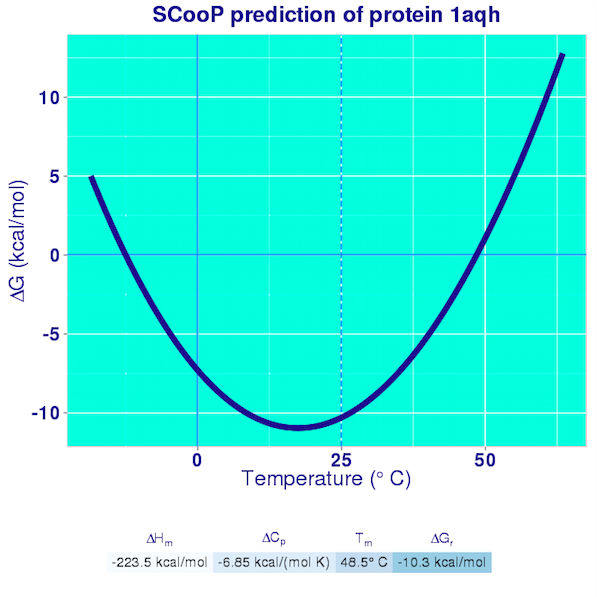
SCooP is very fast and allows a large-scale analysis of the thermodynamic properties of proteins and of the strategies that they use to enhance their thermoresistance. SCooP moreover predicts quite accurately both the thermal stability (defined by the melting temperature) and the thermodynamic stability (given by the folding free energy at room temperature). To the best of our knowledge, no other (physics-based or machine-learning) method is able to compute these quantities in a fast way and to model the dependence of the amino acid interactions on the temperature.

With our conventions, the folded structure is preferred if the value of the folding free energy is negative.
Be aware that non-protein molecules (RNA, DNA, ions, ligands, etc..) are not taken into account in the calculations.
Our prediction method is designed for globular proteins. The stability curve usually diverges at room temperature for membrane proteins or has a bell-shape form with a wrong sign, which indicates the instability of such structures in an aqueous solvent. Only two-state transitions are considered in SCooP and thus, only monomeric structures are considered. In the case of the homodimeric structures, the understanding of the folding cooperativity of the subunits makes the problem even more complicated and we leave it for future analyses. Please contact us for further information. The pH plays an important role in the evaluation of thermodynamic quantities. As SCooP is based on experimental data measured at pH as close as possible to 7, the predicted stability curves are for neutral pH. An extension of SCooP to varying pH values, where the stability curve becomes a 2D surface in 3D space (ΔG, T, pH), is under development. Thanks for visiting us and using SCooP. Do not hesitate to contact us at fapucci@ulb.ac.be, jkwasigr@ulb.ac.be or mrooman@ulb.ac.be. Feedback is most welcome.
Realized by Fabrizio Pucci, Jean Marc Kwasigroch and Marianne Rooman.
3BIO-BioInfo group, CP 165/61, Université Libre de Bruxelles,
50 Franklin Roosevelt Ave, 1050 Bruxelles, Belgium. |
|





