How to submit a query to BRANEart
The first step is to provide the structure of the protein. You may either provide the 4-letter code of the PDB structure, which will then be retrieved from the Protein Data Bank, or upload your own structure file, which must comply with the PDB format. Please note that:The default structure file on the Protein Data Bank does not necessarily correspond to correct quaternary structure of the protein. Select the extension .pdb1, .pdb2, or .pdb3 to use the corresponding Biological Assembly. The format of the "Biological Assembly" files from the Protein Data Bank is somewhat different from the PDB format. In particular, different chains may be referred to as different models of the same chain. In such cases, BRANEart may have to assign new chain names. In case the structure file contains multiple NMR models, only the first one will be considered.
Once the structure file is uploaded/downloaded, BRANEart provides the user with a summary of its content. For each chain, the name (if available) and number of residues is given.
The second step is to define the chains used for the query. Each protein chain present in the structure file can be selected for the computations, or be discarded. Strengths and weaknesses will not be computed on discarded chains, however they will be taken into account for the solvent accessibility computation of selected chains. 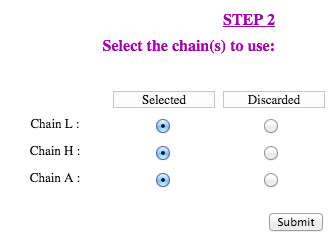
BRANEart Results Upon completion of the second step and submission of the query, BRANEart provides the "Job ID" and a link that has to be followed/bookmarked to check the status of the job. The results will be available on this page as soon as the computations are completed (you may need to refresh the page). They will remain on the webserver for two weeks. For each query, BRANEart reports the following information:A text file containing an index for each residue of the selected chains. This residue index is obtained from a linear combination of a series of statistical mean force potentials derived from transmembrane and extramembrane regions of membrane proteins (see M.N. Mbaye et al, Scientific Reports 9: 12043 (2019) for further details). When the index is close to one, the residue is structurally stable in a lipidic environment. When the residue index is close to zero, the residue is structurally stable in an aqueos environment. A table with the above mentioned index for each residue of the protein coloured according to wheter they are stable in a lipidic (green) or a aqueos (blue) environments. A tool to visualize the protein 3D structure in which each residue is coloured according to the values of its index. The user can zoom on the residue of interest by clicking on it: all atoms including the side chain atoms with their positions are then shown. We have also implemented a series of additional functionalities: it is possible to hide/show a specific chain, select a full screen visualization mode and take a ".png" snapshot of the visualized structures. This page has been tested on Firefox, Google-Chrome, Safari, Edge and Opera. Note that modified residues could not be shown properly.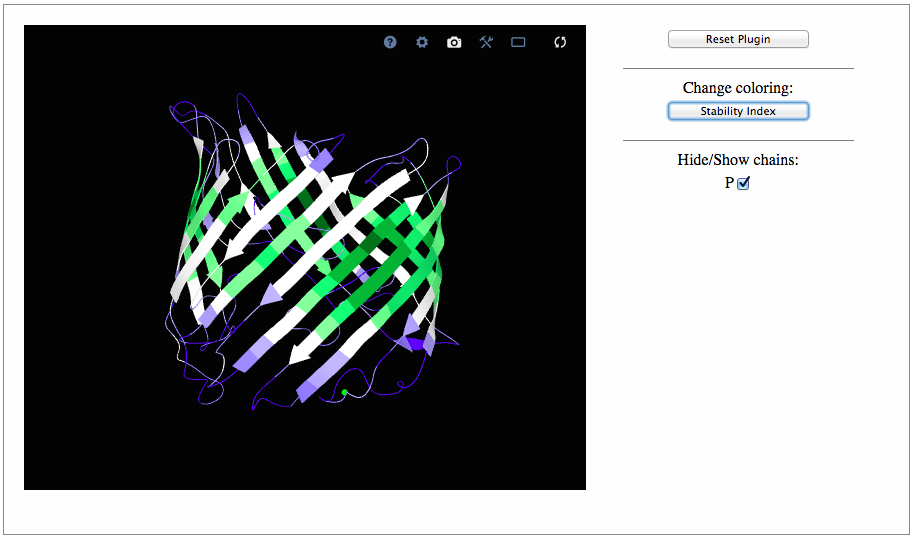
A Pymol session file (.pml) and the PDB are zipped in the file "download.zip" that can be downloaded. It provides the user another visual representation of the residue stability index in the different environments. To use this file, just unzip it and click on ".pml" file to launch the Pymol session. Additional Info The identification of sequence-unique entities relies on the numbering of the amino acids in the structure file. If two chains are identical but numbered differently, they will not be recognized as a sequence-unique entity. In such cases, you may want to edit your structure file and adapt the numbering. This may also be used to introduce a mutation in one chain only and not in other identical chains present in the structure file. In some structure files, chain names are numbers rather than letters. BRANEart does not always deal correctly with this. Try editing your structure file and changing the chain names. Be aware that non-protein molecules (RNA, DNA, ions, ligands, etc..) are not taken into account during the calculations. Note that larger errors should be expected for residues in the vicinity of these non-protein molecules.
| Contacts. Do not hesitate to contact us at mrooman@ulb.ac.be or fapucci@ulb.ac.be. Feedbacks are more than welcome. |
|
|





